



How to transform multi-omic data into knowledge graphs and do useful things with them.
A practical guide to omics data modeling in graphs.

Introduction
Knowledge graphs (KGs) are powerful tools for reasoning and analysis. There is a misconception that KGs only apply to text-based information. The most common objection we hear is that KGs are unsuitable for data science. However, with a few changes, thoughtful data modeling, and novel software, we can unlock the power of these KGs and explore information in data-driven and novel ways.
In this post, I’ll share some frameworks we use at BioBox to craft custom multi-modal knowledge graphs built upon datasets spanning various sequencing technologies. Then, we’ll walk through how this graph is used in the research process to answer questions.
Goals of data integration
Using different experimental techniques and combining data logically can deepen our understanding of biological phenomena. For example, you can use ChIP and ATAC-seq to explore the regulatory control that explains the up/down regulation observed in matched RNA-seq data. Interpreting these datasets requires information about genes, molecular functions, biological processes, etc. Each dataset provides thousands of data points, and the search space is enormous. The goal of data integration is to improve our ability to separate signals from noise.
Data schemas
Metadata capture
The best practice is to isolate all the important variables that can be used for logical flows as nodes in the graph rather than storing them as properties in the node.


Why separate disease and cell type?
It’s simple: we care about the relationships that diseases and cell types have.
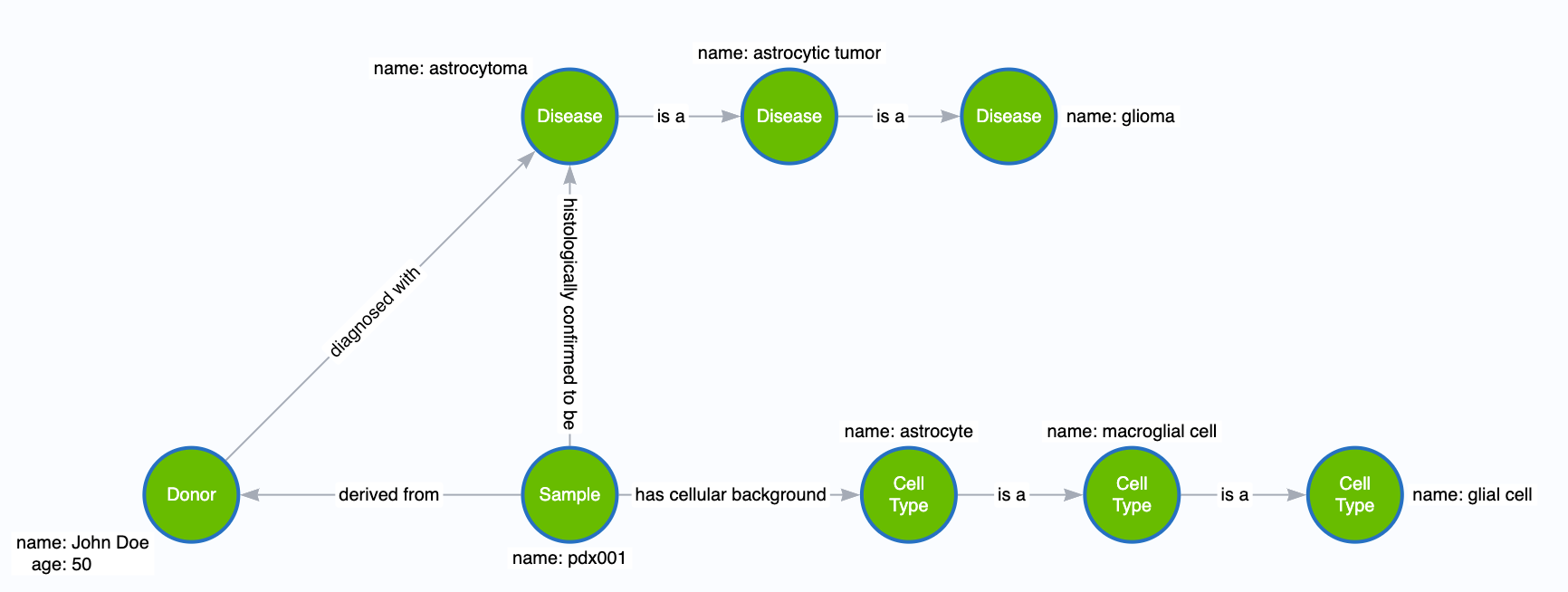
Specifically, we can exploit the ability to walk from semantically related cell types and diseases. By leveraging the power of connected graphs, we can expand our search space for questions like, “Find all glioma samples,” and walk down the paths. Consider what would happen if we stored these values as properties inside the Sample or Donor nodes. We would need to exhaustively search all samples for some key-value match for every value along the semantic chain of values (glioma, astrocytic tumor, astrocytoma), which becomes prohibitively inefficient.
RNAseq
RNA-seq is relatively straightforward to model. These experiments produce two types of datasets: Gene Expression and Differential Expression. You want to capture some information about the transcriptional state of a gene in a biological context (e.g., disease vs. normal, cell population A vs. cell population B, etc.). The gene expression graph mapping is relatively straightforward once the metadata is appropriately set up. We can describe the expression of a gene within a particular RNA-seq library with a direct edge as follows:

Differential gene expression (DGE) datasets require an additional hyper-edge to capture the comparison information.

Because we use a property graph as the base, we can persist the quantitative values of the log2 fold change, p-value statistics, count values, etc., directly inside the edge. It also becomes useful to incorporate these values as edge weights in other graph-based algorithms (more on that in the future). This is a unique characteristic of property graphs and data graphs that differs from traditional RDF-based knowledge graphs.
ChIPseq
Tracking epigenomic observations requires the graph to comprehend genomic coordinates. To do this, we start by creating 1 kilobase (kb) bins across the entire genome and represent them as beads on a chain.
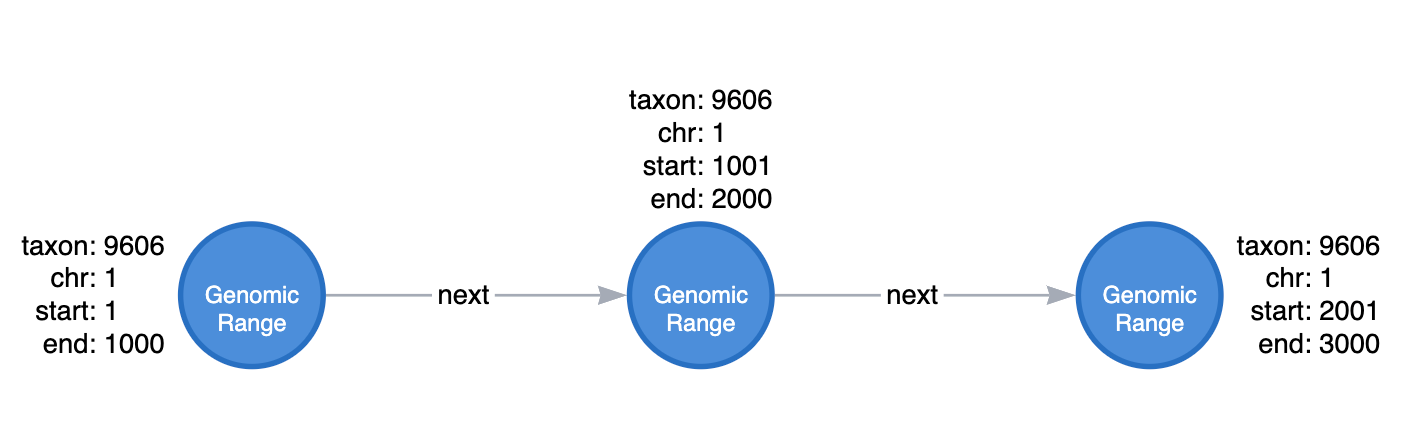
Then, we can load ChIP-seq observations, such as enrichment peaks, into our graph. Each peak is represented as a distinct node with edges to genomic range nodes that demarcate the genomic region from which the peak was detected.

In our graph, we have also loaded regulatory gene features, such as promoters and enhancers. By combining this information, we can compose more complex and meaningful patterns. For example, in the figure above, the topology describes an EP300 peak bound detected to overlap with gene ABC123’s promoter region, suggesting possible epigenetic control.
Doing useful things with multi-omic graphs.
The usefulness of data comes down to whether or not it can impact decision-making. Ultimately, we are hunting for biological explainability so that we can attribute the importance of a target to the overall etiology of a disease. This forms the basis for a drug program to spend millions of dollars around this therapeutic hypothesis. Combining data modalities enhances the ability of scientists to see the bigger picture and make better decisions.
Extending the graph to answer questions
There are only two requirements for graph model construction:
There are two concepts defined (Bipartite graph).
There are relationships in your ontology that can directly or indirectly connect these concepts together.
Types of Bipartite Graphs built on the BioBox Platform:
Gene <> Disease
Gene <> Gene Module
Drug <> Adverse Event
Protein <> Phenotype
Example: Transcriptional Circuits & Pleiotropy
From transcriptomics, we can generate differentially expressed genes and run the count data through GSEA to yield some enriched pathways and gene sets. Typically, we’re hunting for pathways that contribute to an observed phenotype of our disease area. Chances are you will have an overwhelming amount of DE genes and pathway hits. How can we systematically qualify and eliminate targets in a principled way?
One approach is to layer on epigenetic data to identify targets that have the highest impact across multiple dysregulated pathways.
Collect the list of genes that were up-regulated and found in enriched gene modules of interest.
Evaluate the context-specific binding of transcriptional factors (TF) at the gene list promoter regions.
Evaluate the context-specific accessibility of promoter regions for gene list.
Rank the TFs based on occurrence
Together, we are filtering for the strongest transcription factors driving the up-regulation of genes involved in disease pathways/mechanisms. It turns out when the data is modeled and connected in the graph, the context-heavy question is trivial to search for.

In practice, this means we are building a graph model to compute the score between two concepts: Disease and Gene. Each layer of data integrated is an additional piece of information that builds up a scientific hypothesis. Each line of evidence can be used to validate or invalidate a hypothesis to varying degrees. In other words, these lines of evidence can be positively or negatively weighted values. The direction depends on the research question and the hypothesis.
For example, in a target prioritization exercise:
+ 0.3, Gene is up-regulated in disease vs. normal
+ 0.1, Gene participates in disease-specific pathways of interest
+ 0.2, Gene is a TF
+ 0.4, Gene product binds to promoters of disease vs. normal up-regulated genes
- 0.3, Gene is down-regulated in disease vs. normal
- 0.1, Gene product binds to promoters that are not accessible
The bullets above are reachable graph paths in the knowledge graph as defined by the ontology. An instance of this graph path is an idempotent and unique trace. For example, given 10 differential expression datasets, there can be (up to) 10 paths in the graph connecting a disease to a gene.
This is an important difference from traditional knowledge graphs.
On repeated observations, you should probably choose a scaling factor so it doesn’t dominate the overall score. By default, we use harmonic sums, but you’re free to choose whichever method you desire. Summing across positive and negative weighted paths will give you an overall aggregated score to rank objects from these two concepts together.
Conclusion
Crafting a knowledge graph isn’t just about connecting data; it’s about empowering researchers to uncover new insights, make informed decisions, and drive innovation in ways that were previously unimaginable. By integrating diverse data modalities—whether it's transcriptomics, epigenomics, or beyond—we can construct a rich, interconnected tapestry of information that shines a light on the complex biological mechanisms at play.
At BioBox, we've seen firsthand how a well-constructed knowledge graph can transform the research process, turning what was once a daunting sea of data into a navigable map that leads directly to actionable discoveries. Whether you’re prioritizing drug targets, exploring disease mechanisms, or seeking new therapeutic hypotheses, the power of a multi-modal knowledge graph can be the difference between a promising lead and a breakthrough.
But remember, the true value of a knowledge graph lies not in its complexity but in its utility. It’s not just about storing data in a visually appealing structure—it’s about using that structure to generate meaningful results that can drive your research forward.
So, as you consider building your own knowledge graph, ask yourself: How will it serve your team? What questions will it help answer? With the right design and a clear purpose, your knowledge graph could be the key to unlocking the next big discovery in your field.
If you’re ready to harness the power of knowledge graphs in your research, visit us at biobox.io to learn more and connect with our team of experts. Let’s explore the future of data-driven science together.